See a short video explaining on of our main research lines: Engineering the microbiome under individualized perturbations:
MDPbiome - Artificial Intelligence for engineering microbiomes through perturbations
Our algorithm MDPbiome (publication, github) is a methodology for guiding the evolution of a microbiome through successive perturbations, where the algorithm calculates the most likely response of the microbiome to that intervention.
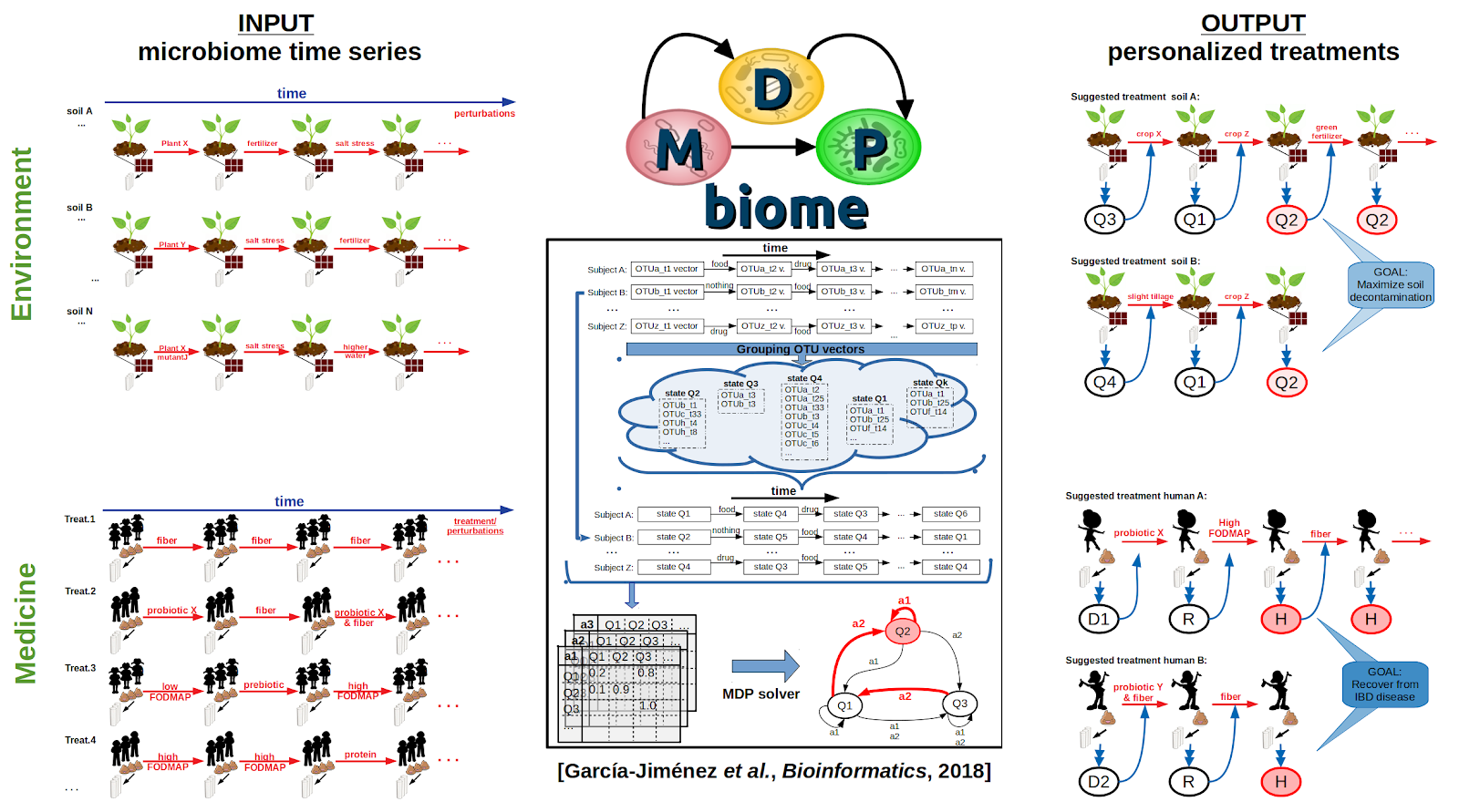
The latest studies on the dynamics of microbiomes highlight that it is currently not possible to predict the effect of a specific external perturbation on a complex microbial community. MDPbiome contributes to addressing this challenge, modeling the effect of perturbations in a microbiome over time as a Markov Decision Process (MDP). Given an initial microbial composition, in any ecological niche or cavity, MDPbiome suggests the sequence of external disturbances that will guide/modulate the microbiome towards a target state, such as a healthier or more performant composition; as well as avoiding undesirable states, such as those associated with a pathology. The study demonstrates the flexibility of MDPbiome as applied to various sets of longitudinal microbiome data, where meta-data regarding disturbances were known (knowledge that is not usually collected and/or published). Measures are also provided to evaluate the performance in terms of reliability and universality of the recommendations proposed by MDPbiome in each case. The potential of MDPbiome will improve in the coming years, as the availability of longitudinal microbiome datasets, and the rich metadata associated with them, increases. Microbial communities associated with plants are also amenable to this approach, to improve their health or nutrition through MDPbiome recommendations, for example, by optimizing soil fertility or proposing low impact policies to develop a sustainable agriculture.
Microbiome states
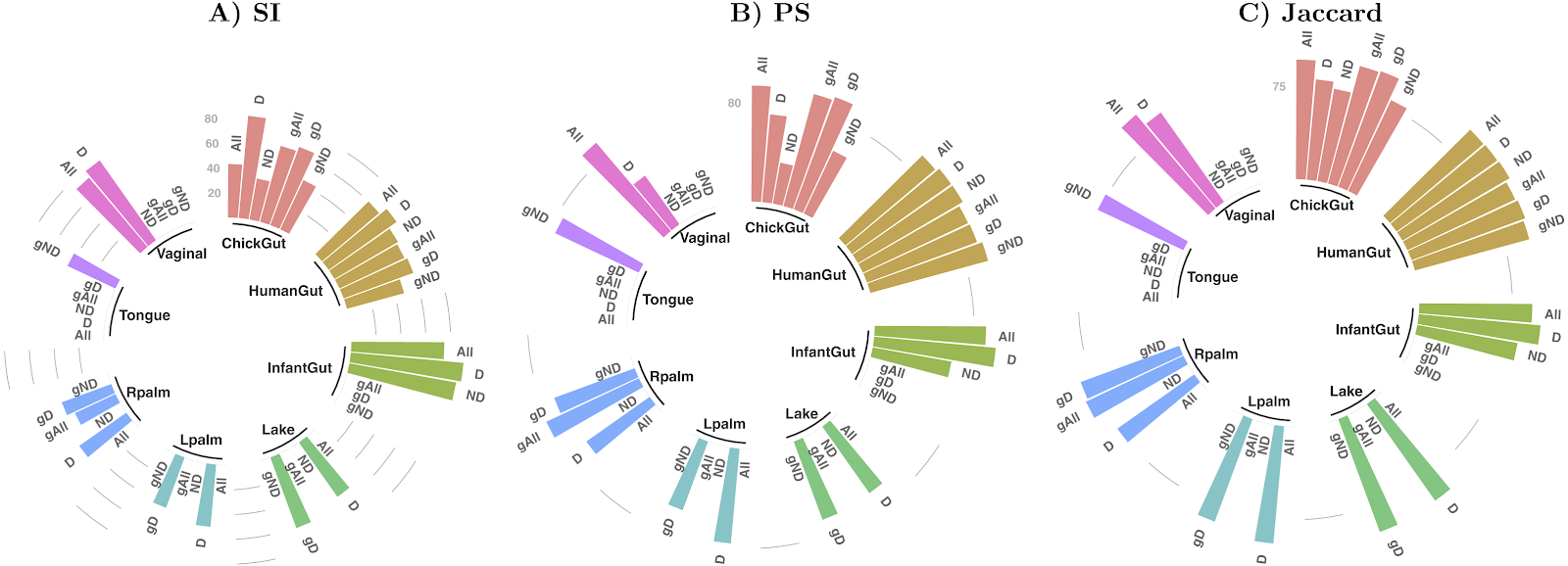
We have defined an automated, generic, objective, domain-independent, and internally-validating procedure to define statistically distinct microbiome states within datasets containing any degree of phylotypic diversity (publication, github). Robustness of state identification is objectively established by a combination of diverse techniques for stable cluster verification. The final output is a set of robustly defined states which can then be used as general biomarkers for a wide variety of downstream purposes such as disease-association studies, monitoring response to intervention, or identifying optimally performant populations.
MDPbiomeGEM - Modeling microbiome personalized treatments, by simulating dynamics under perturbations
[In collaboration with Jorge Carrasco Muriel]
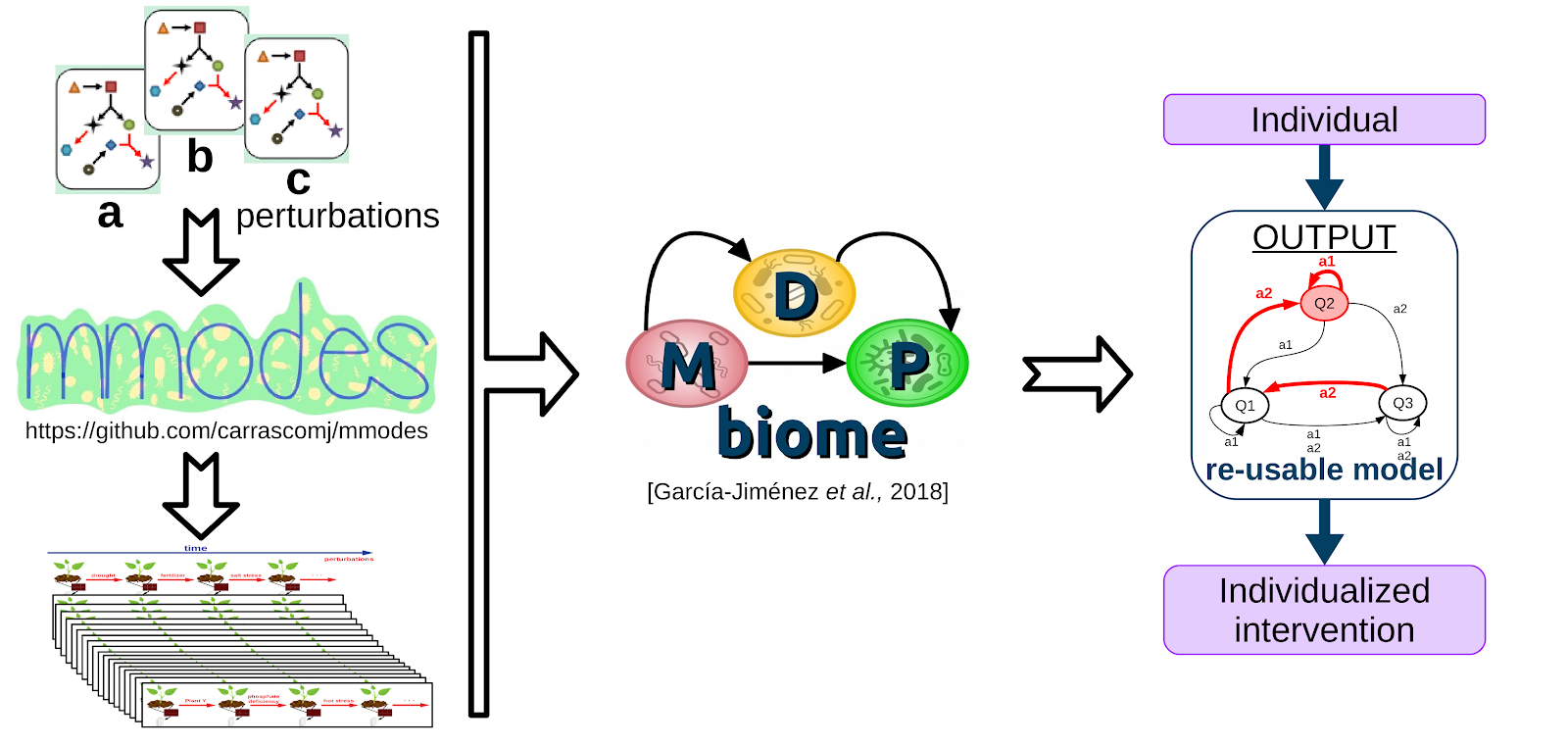
There are few large longitudinal microbiome studies, and fewer that include planned, annotated perturbations between sampling-points. Thus, there are few opportunities to employ MDPbiome to create predictive models, since these are the required input data. Therefore, we develop a novel computational system to simulate the dynamics of microbial communities under perturbations, using genome-scale metabolic models (GEM). Perturbations include modifications to a) the nutrients available in the medium, allowing modelling of, for example, prebiotics; or b) the microorganisms present in the community, to model the consequences of such perturbations as probiotics or pathogen infection. These simulations generate the quantity and types of information which can be used as input to the MDPbiome system. We named this novel combination MDPbiomeGEM(pre-print, github). We demonstrate that MDPbiomeGEM is able to model the influence of prebiotic fiber and probiotics in the case of a Crohn's disease microbiome (conference communication, poster, video talk), or to highlight the relevance of some metabolites in herbicide degradation in soil microbiome. Our system could also contribute to design (perturbed) microbial community dynamics experiments, potentially saving resources both in natural microbiome scenarios by optimizing sequencing sampling, or to optimize in-vitro culture formulations for generating performant synthetic microbial communities.
Deep Learning in microbiome
[In collaboration with Jorge Muñoz from Serendeepia and Sara Cabello Pinedo]
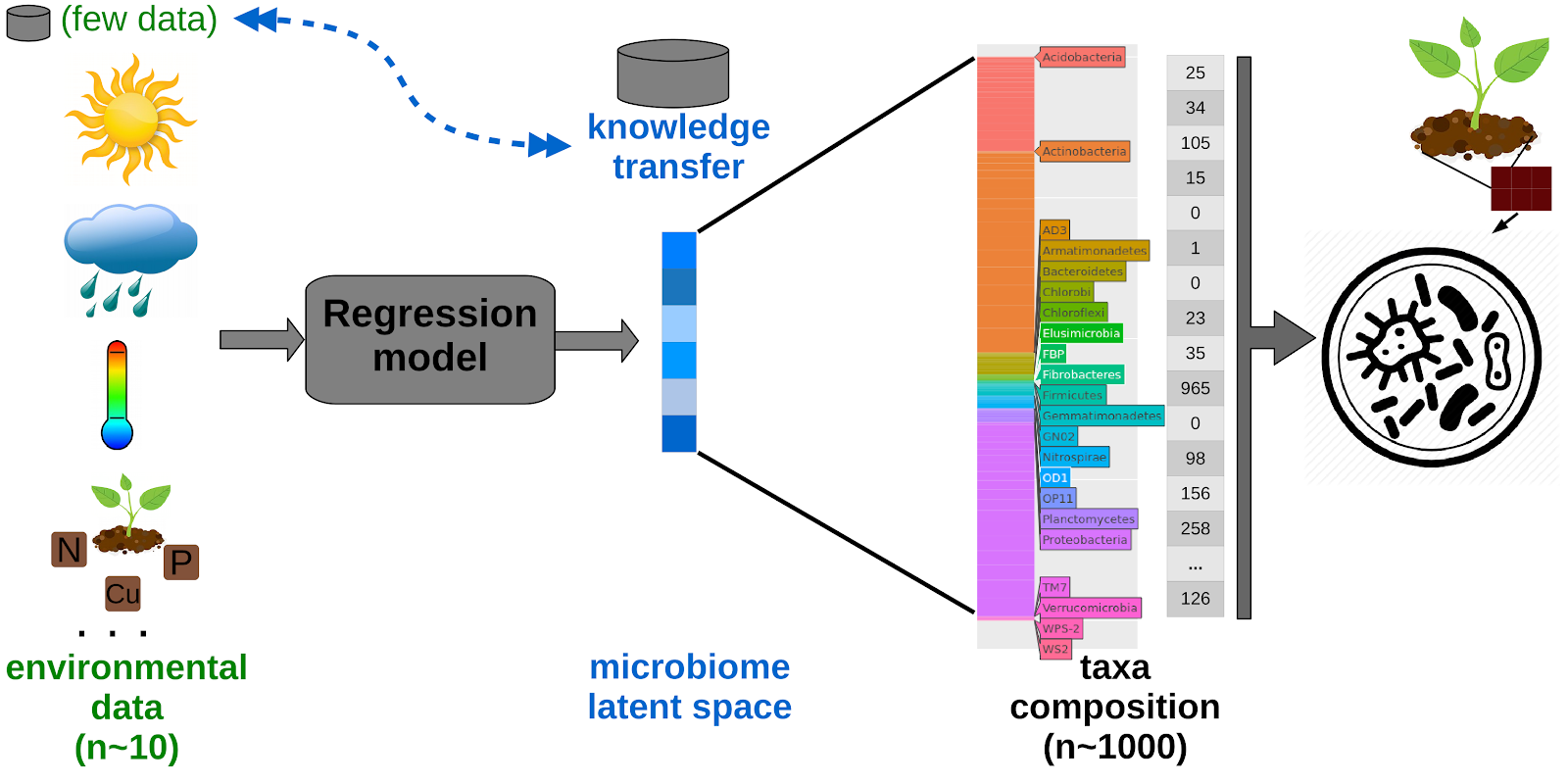
This research line brings together Deep Learning techniques and microbiome data. We have developed a latent space or condensed microbiome representation using transfer and deep learning to promote microbial composition prediction (pre-print, poster, github). We have developed a strategy to reduce the dimensionality of microbiome datasets, such that they can be interrogated and explored more easily. We selected a particular type of artificial neural network - an autoencoder - to condense the long vector of microbial abundances into a short vector (i.e. a latent space) which is more amenable to various kinds of analyses. In particular, we design a model to predict the deep latent space and, consequently, to predict the complete microbial composition using environmental features as input. The performance of our system is examined using the rhizosphere microbiome of Maize.
Analysing microbiome datasets with Machine Learning
[In collaboration with Joaquín Medina (INIA) y el laboratorio de Mauricio González (Universidad de Chile)]
We are developing classification models to discriminate microbiome samples associated with different sample properties. For example, with a large and diverse Atacama desert soil microbiome collection, from different altitudes and with distinct associated plants, our models are able to discriminate microbiome samples between bulk soil versus closer rhizosphere soils, in distinct vegetation belts, using microbiome composition (at different taxonomic levels) and/or nutrients as input.
In addition, we are beginning to apply Deep Learning techniques to reduce dimensionality in maize root microbiome datasets.
We could apply similar prediction approaches to analyse new microbiome datasets, selecting the suitable learning technique, depending on the challenge to solve.


